DNA甲基化会调控基因的表达水平,进而影响基因的相互作用。将基因的相互作用网络和差异甲基化信息结合起来,基于那些甲基化水平发生差异的基因,从整个相互作用网络挖掘出这些基因的相互作用模块,这些模块可以看是与样本表型数据相关的基因集合,这种研究方式叫做Functional Epigenetic Modules(FEMs), 也叫做hotspots。
在ChAMP中,通过champ.EpiMod函数进行FEM分析,用法如下:
> library(ChAMP)
> testDir=system.file(“extdata”,package=”ChAMPdata”)
> myLoad <- champ.load(testDir,arraytype=”450K”)
> myNorm <- champ.norm()
> myEpiMod <-champ.EpiMod(beta=myNorm,pheno=myLoad$pd$Sample_Group)
EpiMod 基于两个输入数据:
- PPI network 蛋白质相互作用网络
在ChAMP中,蛋白质相互网络使用的是别人提供的数据集hprdAsigH.m。 整个网络采用邻接矩阵的表示方式,网络中中每个节点是Entrez Gene ID。
> data(hprdAsigH)
> class(hprdAsigH.m)
[1] “matrix”
> str(hprdAsigH.m)
num [1:8434, 1:8434] 0 1 1 0 0 0 0 0 0 0 …
- attr(*, “dimnames”)=List of 2
..$ : chr [1:8434] “1510” “10436” “7917” “4173” …
..$ : chr [1:8434] “1510” “10436” “7917” “4173” …
需要注意的是,这个数据集是在champ.EpiMod函数中直接定义的,也就是说在不修改源代码的情况下,我们只能基于这个数据集的PPI网络进行分析。
但是通常情况下,我们会从其他数据库中获取到基因的PPI网络,比如STRING数据库,如果要基于STRING数据库的PPI网络进行挖掘,就必须修改源代码了。
2. 差异甲基化信息
首先读取预处理之后的beta matrix和分组信息Sample_Group,然后进行差异分析
PPI和甲基化差异信息准备好之后,就可以基于这两个数据进行EpiMod分析。分析的结果是一个一个的module, 每个module看作是从整个PPI网络中提取出来的sub network。
默认情况下,module的PDF格式的图片保存在工作目录下的CHAMP_EpiMod下,同时还会生成topEpiModLists-Epi-X.txt和topEPI-Epi-X.txt两个文件
CHAMP_EpiMod目录下是所有的module 的PDF 图片
以第一个module ANK2为例,
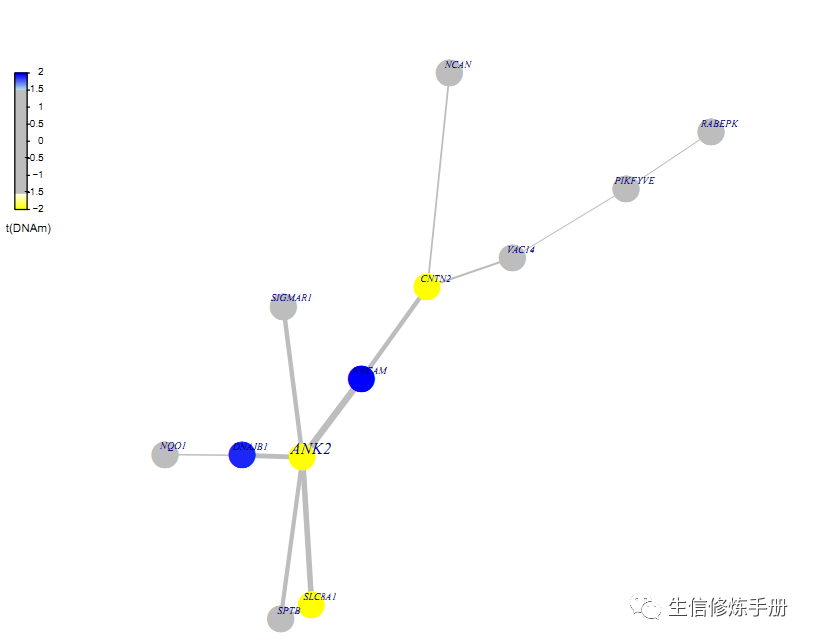
图中的每个节点是一个基因,其相互关系是PPI网络中定义好的,节点的颜色根据差异甲基化的T值定义,小于-1.5的为黄色到白色的渐变色,大于1.5为浅蓝色到蓝色的渐变色,中间的是灰色。
ChAMP只提供了基于差异甲基化信息从PPI网络中挖掘核心module的功能,本值上是通过调用FEM这个R包实现的,在这个R包中,还实现了基于基因水平的差异表达信息从PPI网络中挖掘核心module 的功能,具体的可以查看FEM的帮助文档。










